Лечение синдрома Ретта
В настоящее время нет лекарства от СР, и медицинское лечение направлено на облегчение симптомов у пациентов с помощью мультидисциплинарного подхода. Некоторые из медицинских проблем, которые необходимо решать у пациентов с СР, включают судорожные расстройства, изменения в поведении, нарушения сна, нарушения дыхания, сердечную дисфункцию (удлиненный интервал QT), желудочно-кишечную дисфункцию и переломы костей. Примерно 60% пациентов с синдромом Ретта страдают от некоторых судорожных расстройств. Варианты лечения для облегчения судорог включают вальпроат, ламотриджин, леветирацетам карбамазепин и АЭП. Изменения в поведении чаще всего с тревогой лучше всего бороться с помощью ингибиторов обратного захвата серотонина (СИОЗС). Пациенты с СР часто испытывают трудности с началом сна и частыми ночными пробуждениями, которые можно купировать с помощью надлежащей гигиены сна и тразодона, как только будут исключены обструкции дыхательных путей и другие причины. Нарушения дыхания могут включать апноэ, гипервентиляцию и задержку дыхания. Успешное лечение этих нарушений может быть затруднено
Однако следует соблюдать меры предосторожности, чтобы избегать лекарств, которые изменяют характер дыхания, например, опиоидных препаратов. Нарушения сердечной деятельности включают удлиненный интервал QT
У пациентов с СР это может быть труднее поддаваться лечению по сравнению с общей популяцией. Исследования на мышах с мутациями MECP2 показывают, что бета-блокаторов может быть недостаточно. Следует соблюдать меры предосторожности, чтобы избегать лекарств, которые могут еще больше удлинить интервал QT, например, макролидов. Переломы костей в 4 раза чаще встречаются у пациентов с СР по сравнению с общей популяцией, и уровень витамина D следует тщательно контролировать и дополнять по мере необходимости. Проблемы с пищеварением, такие как гастроэзофагеальная рефлюксная болезнь (ГЭРБ) и запор, часто встречаются у пациентов с СР, и их можно лечить с помощью карбоната кальция, блокаторов гистаминовых Н2-рецепторов (избегайте циметидина) и увеличения потребления клетчатки. Другие варианты лечения включают физиотерапию, логопедию, трудотерапию и психосоциальную поддержку семей. Лечение этих состояний может существенно улучшить качество жизни пациентов с синдромом Ретта, и их не следует упускать из виду.
Дифференциальный диагноз
Синдром Ретта часто может быть неправильно диагностирован. Важными дифференциальными диагнозами, которые следует учитывать, являются церебральный паралич, аутизм, синдром Ангельмана и неспецифическая задержка развития.
Стадирование
До пересмотра критериев СР и открытия MECP2 была внедрена система стадирования в качестве руководства для клиницистов по отслеживанию клинического течения СР
Важно отметить, что система определения стадии не предсказывает ожидаемую продолжительность жизни и не должна использоваться в диагностических целях. Стадия 1 начинается примерно через 6-18 месяцев и включает остановку развития
Некоторые признаки включают ограниченный зрительный контакт, замедление роста головы, неспецифическое заламывание рук и грубые моторные задержки. Стадия 2 начинается в возрасте от 1 до 4 лет и состоит из регресса и быстрого ухудшения. Стадия 2 характеризуется стереотипными движениями рук (отжимание рук или стирка), потерей речи, раздражительностью и нарушением сна. Третья стадия начинается в возрасте от 2 до 10 лет и характеризуется улучшением поведения, навыков общения и использования рук. 4-я стадия начинается в возрасте после 10 лет и характеризуется дистонией, ограниченной подвижностью и брадикинезией.
Прогноз
Ожидаемая продолжительность жизни индивидуума с СР может варьироваться в зависимости от типа унаследованной мутации MECP2. Обычно люди с СР доживают до среднего возраста, и недавние исследования предполагают, что они могут прожить дольше.
Остались вопросы? Задайте их бесплатно нашим врачам в чате ВК ”
” или в .
Беспокоитесь о своем психическом здоровье? Предлагаем пройти диагностику у наших высококвалифицированных психиатров в Москве. Мы работаем строго в соответствии с принципами доказательной медицины, соблюдаем профессиональную этику и поддерживаем высокое качество обслуживания пациентов. При обращении к нам гарантируем полную конфиденциальность без постановки на учет.
Помочь Вам готовы более 50 опытных специалистов центра «Эмпатия». Получить эффективную помощь наших психиатров, психотерапевтов, психологов и эндокринолога можно на очном приёме в Москве и Реутове, а также в формате онлайн-консультации в любой точке мира. Отметим, что выписка рецептов по итогам онлайн-приёма возможна!
Как развивается синдром?
Мозговые функции угасают постепенно, поэтому патология с годами проходит 4 стадии:
- Стагнация. С полугодовалого возраста ребенок теряет активность, появляется апатия к окружающему миру. Снижается количество движений. Отмечается замедление роста, особенно головы.
- Прогрессирующая отрицательная динамика. С полутора до 3-4 лет теряются навыки общения, пропадает речь. Появляется заламывание, приступы нервозности. Отмечаются проблемы с дыханием. К концу периода причиной серьезного беспокойства становятся судорожные припадки.
- Относительное замедление патологических процессов. С 4 до 8-10 лет дети – результат предыдущих аномалий. Они не способны общаться, присутствует серьезная умственная отсталость. Рост идет очень медленно. Ребенка мучают судороги и эпилептические приступы.
- Завершающая стадия. После 10 лет дети с синдромом Ретта меньше страдают от судорог. Появляются серьезные нарушения опорно-двигательного аппарата, значимые отставания в росте головы и тела. Дыхательные процессы неравномерны. Отмечаются патологии внутренних органов.
Далее, то, как будет жить ребенок, зависит от принятых мер по его адаптации, лечению, уходу. Как говорит статистика, женщины с болезнью Ретта проживают более сорока лет. Смерть случается из-за болезней сердца, прободения желудка, прекращения функционирования ствола мозга, судорожных проявлений.
Основные признаки
После 4–5 лет наблюдаются следующие проявления патологии:
- Постепенное замедление роста вплоть до полной остановки.
- Начавшаяся речевая активность затухает, способность общаться пропадает.
- Отставание в психическом развитии.
- Аномалия мышц ступней и ладоней.
- Возможно поредение волос.
- Уменьшение головы и мозга характерны для синдрома, это называется микроцефалией. При этом пропорции остального тела сохраняются.
- Не всегда ребенок может ходить, и даже если такая функция доступна, то только с помощью других людей.
- Тело скованно (гипертонус), расслабленность движения отсутствует при патологии Ретта. Либо, наоборот, мышцы вялые (гипотонус).
- Ребенок не может координироваться в пространстве.
- Непредсказуемые спазмы мышц – дистония. Они заставляют тело резко изменять свою позу.
- Приступы судорог. Эпилептические припадки.
- Патологии позвоночника в виде сколиоза и других нарушений.
- Болезни сердца, сбой ритма.
- Проблемы с органами ЖКТ.
- Лицо, мимика может искажаться от спазма мышц.
- Патологии дыхательной системы. Временная остановка, гипервентиляция легких.
- Бруксизм. Или по-другому ночной скрежет зубов.
- Глаза смотрят в одну точку или блуждают, взгляд расфокусированный.

Сочетание симптомов патологии Ретта различаются в зависимости от конкретного случая, зависят не только от степени поражения мозга, но и сколько предпринято мер по лечению и реабилитации.
Лечение синдрома Ретта
Лечение данного заболевания основано на симптоматической терапии, поскольку невозможно устранить причину развития синдрома и дальнейшее его прогрессирование, однако, можно этот процесс замедлить.
Для того чтобы улучшить состояние ребёнка, применяется медикаментозная терапия препаратами, устраняющими или уменьшающими выраженность некоторых клинических проявлений:
- ноотропные препараты улучшают обучаемость, благоприятно влияют на кровоснабжение головного мозга;
- стимуляторы дофаминовых рецепторов положительно действуют на моторику;
- противосудорожные средства (из-за особенностей заболевания показывают низкую эффективность);
- снотворные.

В зависимости от симптомов, могут быть также назначены лекарственные средства для лечения заболеваний сердечно-сосудистой системы, печени, селезёнки.
Массаж при данном заболевании улучшает мышечный тонус, благоприятно влияет на моторику, оказывает расслабляющее и успокаивающее действие. Лечебная гимнастика и физкультура позволяет улучшить двигательные навыки, поддерживать имеющийся тонус мышц, положительно влияет на эмоциональное состояние больного ребёнка. Психолог и дефектолог помогают улучшить моторику и коммуникативные навыки. Остеопатия положительно действует на состояние позвоночника.
Неплохие результаты показывает музыкотерапия: малыш становится общительнее, уравновешеннее, эмоциональнее, снижается уровень тревожности. Если отсутствует эпилептоидная активность, то может применяться так называемая томатис – терапия, когда ребёнок прослушивает специальную музыку, благоприятно влияющую на его мозговую деятельность. Также применяются иппотерапия (верховые прогулки на лошади), гидротерапия (минеральные ванны, гидромассаж), арт-терапия (творческие занятия отдельно в домашних условиях или в группе музыкой, рисованием и др).

При синдроме Ретта должно соблюдаться лечебное питание. Оно подразумевает диету с повышенным содержанием клетчатки, жиров и витаминов. Ребёнка необходимо кормить высококалорийной пищей и специальными продуктами для увеличения массы тела.
Детям с данным синдромом тяжело пережёвывать еду, содержащую грубые волокна (сырые овощи, фрукты, мясо), поэтому её следует измельчать и давать в виде пюре. Во время кормления необходимо следить за тем, чтобы голова ребёнка находилась под правильным углом и не запрокидывалась назад.

Симптомы Синдрома Ретта:
В анте- и перинатальном периодах, в первом полугодии жизни развитие оценивается как нормальное. Однако во многих случаях наблюдаются врожденная гипотония, незначительное отставание в становлении основных двигательных навыков. Начало заболевания от 4 мес. до 2,5 лет, но наиболее часто оно проявляется в возрасте от 6 мес. до 1,5 года. Описывая психопатологический процесс при синдроме Ретта, одни авторы говорят о «дементировании», другие – о неравномерности психических нарушений.
В течении заболевания выделяют 4 стадии:
I стадия (возраст ребенка 6-12 мес.): слабость мышечного тонуса, замедление роста в длину кистей, стоп, окружности головы.
II стадия (возраст 12-24 мес.): атаксия туловища и походки, машущие и подергивающие движения рук, необычные перебирания пальцами.









III стадия: утрата ранее приобретенных навыков, способности к игре, коммуникациям (в том числе визуальным).
IV стадия: распад речи, возникновение эхолалий (в том числе ретардированных), неправильное употребление местоимений.
Первая стадия – стагнация. Включает замедление психомоторного развития ребенка, замедление роста головы, потерю интереса к играм, диффузную мышечную гипотонию.
Вторая стадия – регресса нервно-психического развития – сопровождается приступами беспокойства, «безутешного крика», нарушениями сна. В течение нескольких недель ребенок утрачивает ранее приобретенные навыки, перестает говорить. Что часто ошибочно интерпретируется как аутизм. Появляются стереотипные движения – «мытье рук», их сжимание, стискивание, сосание и кусание рук, постукивание ими по груди и лицу, атаксия и апраксия. Нарушается равновесие при ходьбе, теряется способность ходить. Больше чем у половины детей отмечается аномальное дыхание в виде апноэ до 1-2 мин, чередующееся с периодами гипервентиляции. Дыхательные нарушения отмечаются в период бодрствования и отсутствуют во время сна. У 50-80% девочек с синдромом Ретта возникают эпилептические припадки различных типов, плохо поддающиеся терапии антиконвульсантами. Чаще всего это генерализованные тонико-клонические припадки, комплексные и простые парциальные судороги, дроп-атаки.
После фазы регресса наступает третья стадия – псевдостационарная, охватывающая длительный период дошкольного и раннего школьного возраста. Состояние детей относительно стабильно. На первый план выступают глубокая умственная отсталость, судорожные припадки, экстрапирамидные расстройства по типу мышечной дистонии, атаксии, гиперкинезов. Приступов беспокойства не отмечается.
В конце первого десятилетия жизни начинается четвертая стадия – прогрессирования двигательных нарушений. Больные становятся обездвиженными, нарастают спастичность, мышечные атрофии, вторичные деформации – сколиоз, появляются вазомоторные расстройства преимущественно в нижних конечностях. Характерно отставание в росте без задержки полового созревания. Имеется тенденция к развитию кахексии. Судорожные приступы редкие. У больных с синдромом Ретта на фоне тотального распада всех сфер деятельности наиболее длительно сохраняются эмоциональное общение и привязанности, соответствующие уровню их психического развития.
Причины возникновения
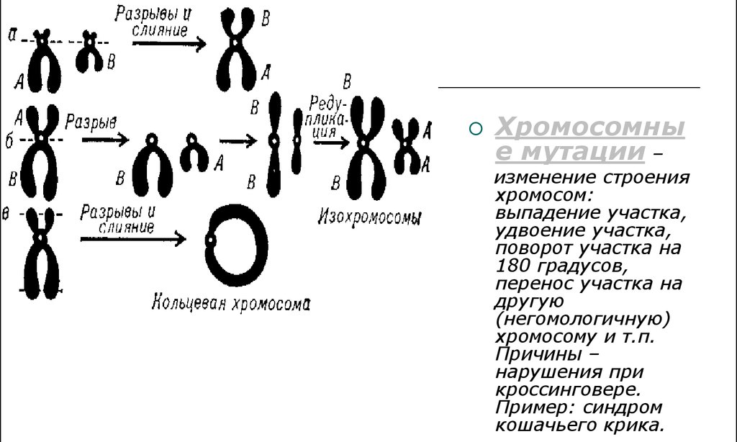
Синдром Ретта у детей в основном встречается у девочек. Это связано с тем, что ген, в котором происходит мутация, вызывающая заболевание, находится в половой Х-хромосоме. У женщин их две, именно поэтому, имея в запасе ещё один нормальный ген, дети женского пола более жизнеспособны. При этом, плод мужского пола, обладая только одной Х-хромосомой с дефектным геном, как правило, погибает ещё внутриутробно. Крайне редкие случаи появления на свет мальчиков с синдромом Ретта сопровождались наличием дополнительной аномалией – так называемым синдромом Кляйнфельтера, то есть они были носителями одной лишней половой Х-хромосомы.
Причины возникновения данного заболевания до сих пор не изучены, несмотря на доказанную наследственную предрасположенность. Существует несколько гипотез, объясняющих механизм данной патологии, но всё же ни одна из них не является единственно верной.
- Генетическая мутация, которая вызвана большим количеством близких родственных связей в родословной. Она возникает ещё в материнской утробе, в период формирования органов и систем плода.
- Хромосомные аномалии. Специалисты сходятся во мнении, что проблемный ген локализируется в половой Х-хромосоме, однако, пока не удалось выяснить, какой именно её участок отвечает за данную болезнь.
- Метаболические нарушения. Некоторые учёные предполагают, что причиной развития патологии могут быть изменения в составе крови: у некоторых больных было обнаружено нетипичное содержание лимфоцитов, а также повышенный уровень пировиноградной и молочной кислоты.
Вполне вероятно, что на развитие синдрома влияют все три фактора, или же в будущем наука откроет новую, пока ещё неизвестную причину его возникновения. Пока достоверно известно только одно: развитие мозга после рождения сильно замедляется, а к четырём годам в черепной коробке у таких больных происходят необратимые изменения, приводящие к остановке роста, атрофии мышц, нарушению работы внутренних органов и эпилепсии.
По некоторым наблюдениям более 70% случаев возникновения синдрома Ретта вызваны нарушениями в генетическом материале отца ребёнка.
Критерии диагностики
Диагноз синдрома Ретта основывается на распознавании характерной клинической картины. Для этого Международной ассоциацией по изучению синдрома Ретта предложена группа диагностических критериев, которые разделены на необходимые, дополнительные и исключающие. Классическая форма синдрома Ретта может быть диагностирована, если у пациента присутствуют все необходимые критерии. Следует отметить, что женский пол не входит в их число, поскольку это могло бы уклонить врачей от поиска мальчиков с синдромом Ретта. Вторая группа состоит из дополнительных критериев, многие из которых обычно имеются у больных, но ни один из них не является обязательным для постановки диагноза. Третья группа — исключающие критерии, одного из которых достаточно, чтобы отвергнуть синдром Ретта у пробанда.
Диагностические критерии синдрома Ретта (по Trevathan et al., 1998) включают необходимые критерии, среди которых нормальные пренатальный и перинатальный периоды, нормальная окружность головы при рождении с последующим замедлением роста головы между 5 месяцами и 4 годами; потеря приобретенных целенаправленных движений рук в возрасте от 6 до 30 месяцев, связанная по времени с нарушением общения; глубокое повреждение экспрессивной и импрессивной речи и грубая задержка психомоторного развития; стереотипные движения рук, напоминающие выжимание, стискивание, хлопки, “мытье рук”, потирание, появляющиеся после потери целенаправленных движений рук; нарушения походки (апраксии и атаксии), выявляющиеся в возрасте 1 — 4 лет. Диагноз считается предварительным до двух-пятилетнего взраста.
Хотя исследователи единодушны в том, что в развитии патологии наследственные факторы играют существенную роль, их мнения относительно механизмов наследования синдрома Ретта расходятся
Дополнительные критерии включают дыхательные расстройства в виде периодических апноэ во время бодрствования, перемежающихся гипервентиляцией, форсированного изгнания воздуха и слюны, аэрофагии; судорожные припадки; спастичность, часто сочетающуюся с дистонией и атрофией мышц; периферические вазомоторные расстройства, сколиоз, задержку роста, гипотрофичные маленькие ступни, электроэнцефалографические аномалии (медленный фоновый ритм в состоянии бодрствования и периодическое замедление ритма (3-5 Гц), эпилептиформные разряды без или с наличием клинических судорог).
Наконец, к исключающим критериям относят очевидность внутриутробной задержки роста, органомегалию или другие признаки болезней накопления, ретинопатию или атрофию дисков зрительных нервов, микроцефалию при рождении, доказательство перинатально приобретенного повреждения мозга, существование идентифицированного метаболического или другого прогрессирующего неврологического заболевания и приобретенные в результате тяжелой инфекции или черепно-мозговой травмы неврологические нарушения.
У ряда больных клинические признаки не соответствуют полностью классическому течению синдрома Ретта. Эти случаи классифицируют либо как неполные, либо как атипичные формы заболевания. При неполной форме у больного присутствуют многие, но не все из необходимых симптомов. Этим характеризуются легкие варианты болезни. Атипичные формы — это случаи синдрома Ретта, которые соответствуют всем необходимым критериям диагностики, но имеют отклонения от типичного течения. В частности, при атипичной форме синдрома с ранним началом судорог эпиприпадки являются дебютом заболевания. Раннее начало эпилепсии не оказывает существенного влияния на течение и прогноз болезни, однако вызывает дифференциально-диагностические трудности. При атипичном варианте синдрома с частично сохраненной речью больные имеют некоторые речевые навыки, течение заболевания у них более мягкое, чем при классической форме, а уровень общения значительно выше. Известны также атипичные варианты синдрома с аномальным развитием ребенка с рождения, поздним началом фазы регресса, сюда же относят случаи синдрома Ретта у мальчиков.
Проведенные в отделе врожденных и наследственных заболеваний МНИИ педиатрии и детской хирургии исследования продемонстрировали значительный клинический полиморфизм синдрома Ретта. Среди 40 наблюдавшихся пациентов в возрасте от 20 месяцев до 16 лет выявлено 27 случаев классического синдрома Ретта и 13 атипичных случаев. Генеалогический анализ показал накопление в родословных пробандов случаев умственной отсталости, судорожных состояний, психических заболеваний. Эти наблюдения, по-видимому, могли являться случаями синдрома Ретта с низкой экспрессивностью.
Симптомы
Беременность и роды мам, у которых дети имеют синдром Ретта, обычно были нормальными. Возраст появления симптомов, их тяжесть и связанная с этим инвалидность широко варьируются среди разных людей.
Однако, похоже, что большинство детей с синдромом Ретта растут и ведут себя нормально в течение первых шести месяцев. После этого появляются признаки и симптомы.
Наиболее выраженные изменения обычно наблюдаются между 12 и 18 месяцами, внезапно, или через период недель или месяцев.
Признаки
Задержка роста.
Рост мозга замедляется после рождения. Голова меньшего размера, чем обычно (микроцефалия), обычно является первым признаком того, что у ребенка синдром Ретта. По мере взросления становится очевидной задержка роста других частей тела.
Потеря нормальных движений, координации.
Первые признаки -уменьшение контроля рук и способности ползать, ходить нормально. Потеря возможностей происходит постепенно. Со временем мышцы ослабевают, становятся жесткими, спазматическими, с ненормальными положениями, движениями.
Потеря коммуникационной способности.
Дети с синдромом Ретта часто начинают терять способность говорить, открывать глаза и общаться другими способами. Они могут потерять интерес к другим людям, игрушкам, к окружению.
Некоторые дети быстро получают изменения, такие как внезапная потеря речи. Со временем большинство, постепенно обретают визуальный контакт, развивают невербальные навыки общения. 
Аномальные движения рук.
Дети с синдромом Ретта часто совершают повторяющиеся и бессмысленные движения руками, которые у каждого разные.
Некоторые скручивают руки, сжимают, трясут, хлопают, бьют ладонями.
Странные движения глаз.
Дети с синдромом Ретта склонны делать странные движения глаз, например, смотреть, моргать, пересекать глаза или закрывать один глаз.
Респираторные проблемы
Некоторые дышат ненормально быстро (гипервентиляция), выдыхают воздух или слюну силой, глотают воздух. Эти проблемы, как правило, происходят во время бодрствования.










Поведенческие
Ажитация и раздражительность.
Дети, страдающие синдромом Ретта, становятся более возбужденными, раздражительными по мере взросления. Периоды крика начинаются внезапно, без видимых причин, и могут длиться часами.
Другое аномальное поведение
Некоторые показывают странное и неожиданное выражение лица, длительные эпизоды смеха, лижут руки, тянут волосы, одежду.
Когнитивные нарушения.
Потеря навыков сопровождается потерей интеллектуального функционирования.
Неврологические
- Судороги. Большинство страдают от судорог в какой-то момент своей жизни.
- Аномальная кривизна позвоночника (сколиоз).
Сколиоз распространен. Обычно появляется от 8 до 11 лет, увеличивается с возрастом. Хирургия может потребоваться, если кривизна слишком суровая.
Нерегулярные сердечные сокращения.
Это опасная для жизни проблема для многих детей и взрослых, страдающих синдромом Ретта, и может вызвать внезапную смерть.
Боль.
Из-за проблем со здоровьем повышен риск боли. Но трудности общения мешают распознавать проблемы, связанные с болью.
Другие симптомы
Может появиться большое разнообразие симптомов, таких как тонкие, хрупкие кости, подверженные переломам; короткие руки и ноги, которые обычно холодны; проблемы жевания и глотания; бруксизм. Симптомы варьируются от одного ребенка к другому.
Что провоцирует / Причины Синдрома Ретта:
Подтверждена наследственная природа заболевания. Вопросы патогенеза заболевания остаются спорными. Генетическая природа связывается с ломкой Х-хромосомой и наличием мутаций в генах – регуляторах процесса репликации. Выявлены селективный дефицит ряда регулирующих рост дендритов белков, низкое количество глутаминовых рецепторов в базальных ганглиях, дофаминергических рецепторов в хвостатом ядре, нарушения холинергической функции. Гипотезу «прерванного развития», в основе которой лежит дефицит нейротрофических факторов, выдвинул D. Armstrong. Предполагается поражение нижних моторных нейронов, базальных ганглиев, вовлечение спинного мозга, ствола и гипоталамуса.
Анализ морфологических изменений при синдроме Ретта указывает на замедление развития мозга после рождения и остановку его роста к 4-летнему возрасту. Выявлено замедление роста тела и отдельных органов (сердца, печени, почек, селезенки).
Симптомы заболевания
Главным проявлением синдрома Туретта у детей являются разнообразные тики. Термин «тик» пришёл к нам из французского языка и буквально переводится, как «подёргивание». При неврологическом заболевании возникновение их происходит постепенно, более простые движения заменяются сложными, появляются новые симптомы.
Разновидности тиков при синдроме Туретта
двигательные.
Начинается болезнь обычно с возникновения простого двигательного (моторного) тика. Чаще всего патологический процесс охватывает мышцы лица и шеи, возникают мигания век, подёргивания мышц, наморщивание носа и другие проявления. С течением времени процесс переходит на мышцы рук (сгибание, выворачивание кистей), плечевого пояса (спазмы мышц туловища), возникают сложные тики с использованием посторонних предметов (указывание шариковой ручкой, непристойные жесты).
При распространении патологии на нижние конечности наблюдаются топтания на месте, подпрыгивания, приседания. Иногда тики носят угрожающий здоровью ребёнка характер – удары головой о стену, сильное прикусывание губ;
вокальные.
После возникновения простых моторных тиков у детей больных синдромом Туретта обычно появляются гиперкинезы мышц нёба, гортани и языка. Малыш начинает издавать нечленораздельные звуки, похожие на лай собаки, шипение, повторение слогов, попёрхивание, покашливание и другие.
Нередко необычность неврологических проявлений приводит к ошибкам диагностики. Вокальные тики вплетаются в поток речи и воспринимаются родителями, как заикания, а навязчивые покашливания длительно и безрезультатно лечатся ЛОР-врачами.
Сложные тики проявляются во внезапном выкрикивании слов или предложений, иногда повторении услышанного (эхолалия). Самый известный симптом – бесконтрольное употребление нецензурных, бранных слов (копролалия), встречается по разным данным в 10 – 30 % случаев заболеваний и не является обязательным для постановки диагноза.
Иногда тики возникают у абсолютно здоровых детей. Обычно это происходит в возрасте от 7 до 12 лет и связаны с незрелостью нервной системы, увеличением психологической и эмоциональной нагрузки. Родители должны обязательно обратиться к врачу в случае усиления проявлений, появления множественных тиков, нарушения качества жизни ребёнка, социальной адаптации. Любой тик, который не проходит самостоятельно более 1 месяца, является показанием к углублённому обследованию малыша.

Особенность течения заболевания
Тики могут возникать при различных неврологических заболеваниях, но при синдроме Туретта проявления этого симптома имеют свои закономерности:
- начало проявлений в возрасте 2 – 5 лет, преобладание пациентов мужского пола;
- сочетание моторных и вокальных тиков;
- огромное многообразие проявлений;
- изменение тиков с течением времени, появление новых более сложных симптомов или самостоятельное регрессирование к окончанию подросткового возраста;
- усиление проявлений при стрессе, эмоциональных перегрузках;
- периоды ремиссии, исчезновения тиков, могут длиться от месяца до нескольких лет;
- внезапное возникновение подёргиваний после периода ремиссии, без видимых на то причин;
- проявление гиперкинезов (избыточных движений) обычно уменьшается во время сна и ранним утром;
- возможность некоторыми пациентами контролировать тики;
- сочетание болезни Туретта с синдромом дефицита внимания, обсессивно-компульсивным расстройством.
Многие дети рассказывают об определённых физических ощущениях перед появлением тика, явлениях першения в горле, зуда, рези в глазах, ощущении инородного предмета в горле. Таким образом ребята понимают, что приближается неприятный симптом и пытаются его контролировать. После подавления гиперкинеза часто возникает чувство дискомфорта, внутреннее напряжение, желание целенаправленно воспроизвести тик.

Выраженность проявлений можно оценить по Шкалы Тяжести Синдрома Туретта (TSSS):
лёгкая степень.
Малыш легко контролирует симптомы недуга, болезнь не заметна для окружающих и не влияет на качество жизни малыша;
умеренные проявления.
Тики не всегда удаётся контролировать, окружающие знают о болезни ребёнка. Возможны проблемы в обучении, взаимоотношениях со сверстниками;
тяжёлые проявления.
Сложные, многообразные моторные и вокальные тики резко затрудняют коммуникацию ребёнка. Отсутствие контроля над происходящим сильно снижает качество жизни малыша.










Прогноз
Синдром Ретта – это генетическое заболевание, поэтому вылечить его невозможно. В настоящее время ведутся активные разработки стратегии применения стволовых клеток, которые будут использоваться для лечения этого недуга. Уже успешно опробованы предварительные средства на лабораторных мышах, что дает надежду и положительный прогноз на скорое открытие эффективного препарата для лечения синдрома Ретта.
Ну а пока ребенок с таким диагнозом должен быть окружен заботливыми и любящими родителями, которые, если помогут ему сохранить приобретенные навыки или научат его основам самообслуживания, то дадут ему возможность прожить до 50 лет и более. Тем более такие случаи уже известны медицине.
Таким образом, синдром Ретта – редкая и тяжелая генетическая патология, развивающаяся обычно у девочек практически сразу же после рождения. Этот недуг затрагивает самые важные участки организма и к 10-летнему возрасту ребенок становится совершенно беспомощным. Эффективного лечения синдрома еще не создано, но разработки в этой области уже ведутся и весьма успешно.
Немного истории
Частота синдрома Ретта широко варьирует. В большинстве европейских государств она составляет 1:10000 — 1:15000 девочек, являясь следующей по частоте после синдрома Дауна специфической причиной тяжелой умственной отсталости у девочек. Проведенные нами исследования позволили установить, что удельный вес синдрома Ретта среди умственно отсталых девочек в России составляет 2,48%.
Многие авторы отмечают, что в одних областях синдром Ретта встречается чаще, чем в других. Были выделены небольшие районы, в основном сельские, которые получили условное название “Ретт-ареалов”. В частности, в одной из провинций Северной Италии частота синдрома Ретта составляет 22:10000 девочек. Относительно большая частота заболеваемости отмечена также в Норвегии, Албании, Венгрии и может быть связана с еще существующими популяционными изолятами .
Несмотря на то что случаи заболевания обычно спорадические, описано не менее 15 семей, в которых синдром Ретта наблюдался у двух и более женщин (тети и племянницы, родных сестер, полусестер по материнской линии). В одной бразильской семье заболевание было диагностировано у трех родных сестер. Следует отметить факт рождения у молодой женщины с синдромом Ретта дочери, которая нормально развивалась до 1,5-летнего возраста, когда постепенно появились клинические признаки синдрома. Убедительным доказательством генетической природы заболевания является то обстоятельство, что в парах монозиготных близнецов с синдромом Ретта всегда поражены обе девочки. С другой стороны, не известно ни одного примера, чтобы в паре дизиготных близнецов оба ребенка страдали синдромом Ретта.
На основании результатов уникального генеалогического исследования, охватившего 128 шведских семей, O. Акессон и соавторами (1995) была предложена новая гипотеза о существовании стартовой премутации при синдроме Ретта. Были составлены родословные, которые включали от 7 до 10 поколений, прослеженных с помощью церковных записей вплоть до начала 18 века. Около половины пробандов имели общее происхождение из одних и тех же небольших сельских районов и даже фермерских усадеб. Оказалось, что у многих больных имеется общий предок, и некоторые поначалу независимые родословные были объединены. Выявленное авторами общее происхождение ряда пораженных детей предполагает передачу премутации, которая через поколения может привести к полной мутации и синдрому Ретта как ее результату. В связи с этой гипотезой особенно перспективны молекулярно-генетические исследования с целью определения увеличения числа повторов нуклеотидных триплетов в ДНК генома при синдроме Ретта.
Синдром Маркуса-Гунна: причины офтальмопатологии
Проводя диагностические мероприятия, врач в первую очередь устанавливает природу синдрома Маркуса-Гунна. Причины его бывают врожденными и приобретенными. При этом, когда речь идет о врожденном характере патологии, не подразумевается наследственность. Генетически заболевание никак не обусловлено. Развивается оно спорадически, то есть случайно. Происходит это еще в период внутриутробного развития. Синдром возникает, если формируется аномальное соотношение между тройничным нервом и нервом, который регулирует движения глазного яблока. Факторы, провоцирующие аномалию, точно неизвестны.

Иногда пальпебромандибулярная синкинезия становится следствием родовой травмы. Однако данная форма патологии уже не врожденная, а приобретенная. Помимо родовой травмы, ее причинами могут стать:
- естественное старение организма, сопровождающееся ослаблением мышц, которые отвечают за поднятие век;
- диабетическая невропатия — осложнение при сахарном диабете, характеризующееся повреждением нервных волокон;
- рассеянный склероз;
- синдром Горнера — поражение симпатической нервной системы, проявляющееся на глазах;
- инсульт;
- черепно-мозговая травма;
- глазная мигрень;
- воспалительные болезни головного мозга, включая энцефалит;
- хроническая недостаточность кровообращения — медленно прогрессирующая дисфункция головного мозга;
- сильные потрясение психологического характера;
- побочный эффект, осложнение после процедуры введения ботокса.